Between April 2021 and March 2024, EFORT has been involved as co-leader in an EU-funded project called CORE-MD.
CORE–MD objectives were to systematically review methodologies for the clinical investigation of high-risk medical devices (Work Package 1), recommend how new trial designs can contribute (Work Package 2), and advise on methods for aggregating real-world data from medical device registries with experience from clinical practice (Work Package 3). Multidisciplinary workshops on 20 and 21 November 2023 proposed a hierarchy of levels of evidence from clinical investigations; educational and training objectives for all stakeholders, to build expertise in regulatory science in Europe; and an ethics charter for medical device innovation (Work Package 4).
Each task and work package leader has successfully coordinated complex and interdisciplinary work streams that has led to the production of deliverables and also to a series of additional spin-off/secondary studies that have further enriched the breadth of the CORE-MD scientific findings and its potential to influence policy making and help address MDR implementation hurdles.
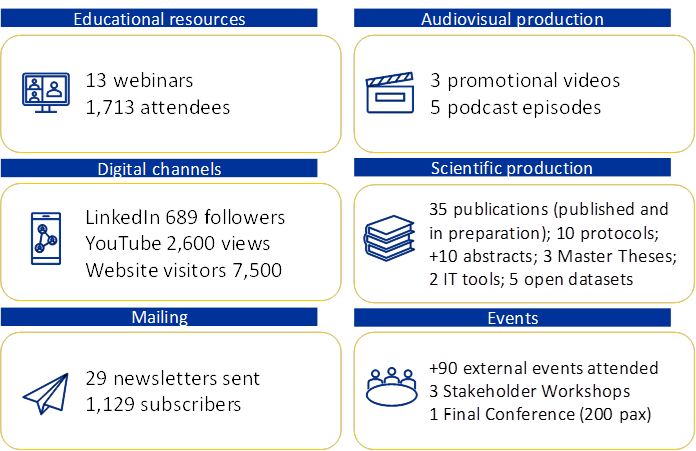
See CORE-MD impact in a nutshell
The project has definitely made bold steps towards this accomplishment by sharing regular updates on its progress with Medical Device Coordination Group (MDCG) and its Clinical Investigation and Evaluation (CIE) sub-group, as testified by the participation of project’s representatives in 14 meetings of MDCG, its subgroups and task forces. Moreover, the CIE Working Group has officially established a dedicated task force (‘Work Package 28’) to carefully review all CORE-MD findings and deliverables and seek the most suitable ways to leverage and integrate them in the next revision of the MEDDEV clinical evaluation guidance or otherwise within the EU regulatory system for high-risk medical devices.
The exploitable outputs (tools, methods, frameworks, and recommendations) co-produced by the multidisciplinary team involved in CORE-MD constitute its tangible legacy to the broader European regulatory science community. These results will be exploited beyond the project’s life. Among the many results, CORE-MD developed a risk scoring system for Artificial intelligence Software, a statistical tool to calculate the risk of a device, a decision framework for post-market surveillance, an ethics charter, etc. For each result, the consortium envisaged an exploitation route.
The contribution of CORE-MD to the implementation of the Medical Device Regulation was significant. Regulators, academics, patients, healthcare providers and professionals, industry and notified bodies have experienced through CORE-MD how constructive dialogue could contribute to a mutual understanding of the needs and expectations of each key player in the field of medical devices.
Many recommendations stemming from the consortium will substantially help regulators to understand better the challenges and the responses to contribute to the consistent and smooth implementation of the MDR. Ultimately, this will help manufacturers navigate all factors involved in bringing high-quality, researched-backed products to market.

Read the CORE-MD Final Conference Booklet
on Regulatory science for high-risk medical devices in the EU


